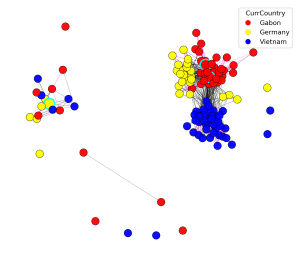
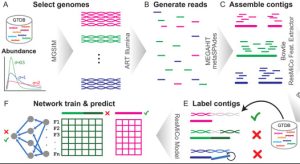
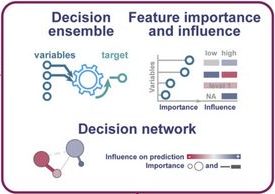
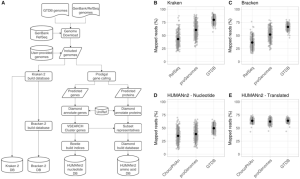
The number of published metagenome assemblies is rapidly growing due to advances in sequencing technologies. However, sequencing errors, variable coverage, repetitive genomic regions, and other factors can produce misassemblies, which are challenging to detect for taxonomically novel genomic data. Assembly errors can affect all downstream analyses of the assemblies. Accuracy for the state of the art in reference-free misassembly prediction does not exceed an AUPRC of 0.57, and it is not clear how well these models generalize to real-world…