In our new publication in Environmental Microbiology, we explore the use of genome-level phylogenies and also genome-derived functional information for assessing alpha and beta diversity. Given the large number of microbial genomes that are now available, we sought to assess how this information can be utilized to gain new insights into microbial diversity. We show that this “genome-based diversity” approach can lead to new understanding of human gut microbiome diversity and can also be quite helpful for classifying metagenomes via machine learning.
Youngblut, Nicholas D., Jacobo de la Cuesta-Zuluaga, and Ruth E. Ley. 2022. “Incorporating Genome-Based Phylogeny and Functional Similarity into Diversity Assessments Helps to Resolve a Global Collection of Human Gut Metagenomes.” Environmental Microbiology, January. https://doi.org/10.1111/1462-2920.15910.
The work presented in our new manuscript was motivated by our recent Struo and Struo2 publications, in which we developed (and then improved) a pipeline for the creation of custom reference metagenome profiling databases with data from the Genome Taxonomy Database (GTDB).
Cuesta-Zuluaga, Jacobo de la, Ruth E. Ley, and Nicholas D. Youngblut. 2020. “Struo: A Pipeline for Building Custom Databases for Common Metagenome Profilers.” Bioinformatics 36 (7): 2314–15.
Youngblut, Nicholas D., and Ruth E. Ley. 2021. “Struo2: Efficient Metagenome Profiling Database Construction for Ever-Expanding Microbial Genome Datasets.” PeerJ 9 (September): e12198.
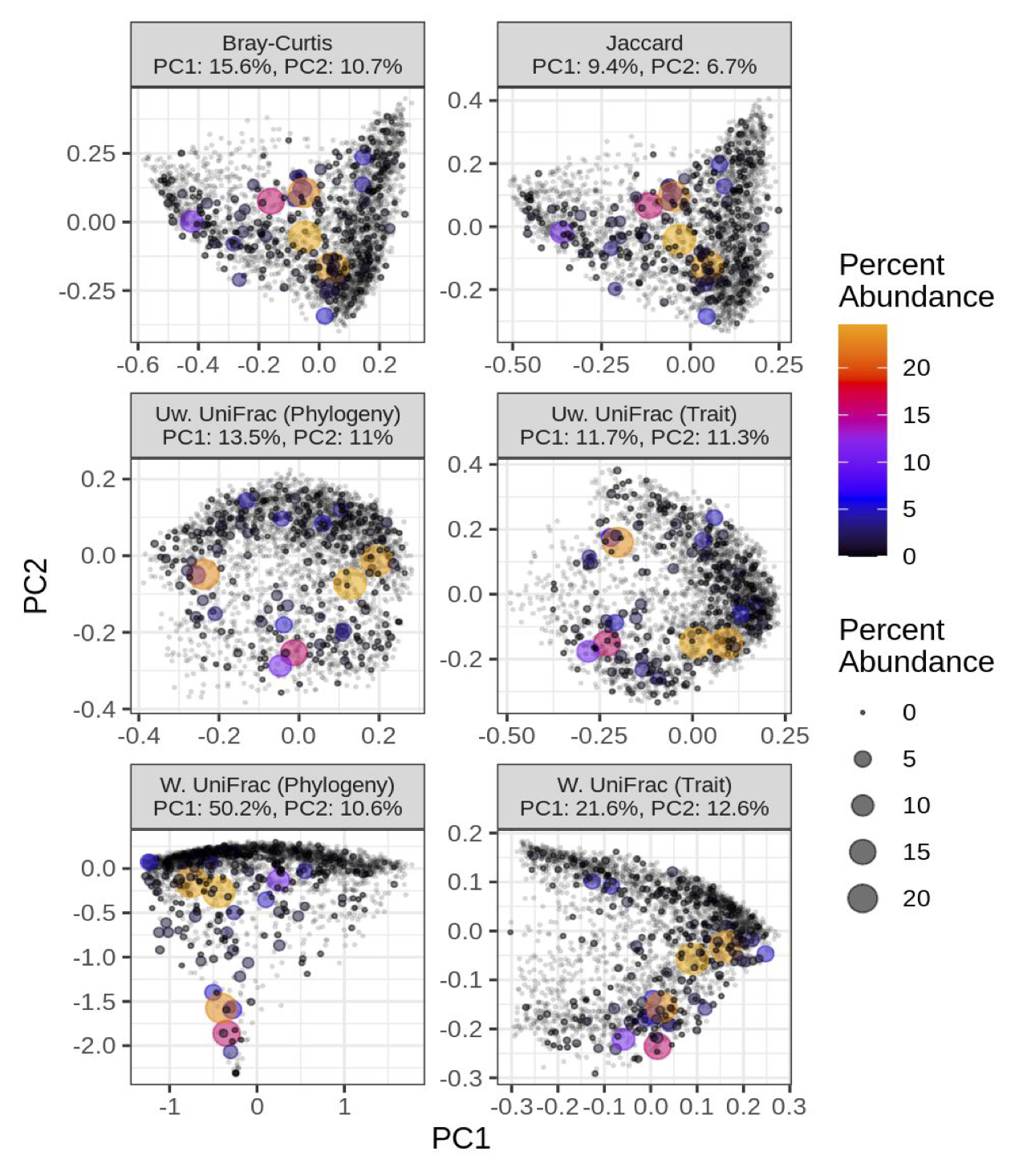
The GTDB includes a multi-locus genome phylogeny for bacteria and archaea, since the entire database is based on genomes, unlike databases such as SILVA or Greengenes. So, we could use our metagenome profiling database to estimate species-level abundances in metagenomes, and importantly, we now had a genome phylogeny representing all species.
This opened the door for utilizing phylogenetic measures of diversity such as Faith’s PD and UniFrac. These measures have been used very effectively for 16S rRNA amplicon-based studies but rarely used for shotgun metagenomics. This disparity stems from the fact that a phylogeny can be directly inferred from 16S rRNA amplicon sequences or by using a pre-existing phylogeny (e.g., SILVA full-length 16S tree); however, shotgun metagenomics targets the entire genome, so creating a single or multi-locus phylogeny directly from the short Illumina reads is more challenging.
Besides a robust phylogeny, genome references include functional information, such as the gene and metabolic composition unique to each genome. Function does not always track phylogeny due to recombination and convergent evolution, so we wanted to see if functional information could provide unique insight into microbiome diversity. To make this functional analysis directly comparable to our phylogeny-based approach, we created “functional similarity trees”, similar to evolutionary relatedness depicted by the genome phylogeny. There are many ways to define genomic “function”, so we utilized COG, Pfam, and traits (e.g., aerobic versus anaerobic growth) inferred via machine learning from select Pfam domains.
For both alpha and beta diversity, our tree-based diversity measures (phylogeny and function) provided unique insight into how microbial diversity is partitioned across a large, global collection of human gut metagenomes (2943 samples). For instance, we compared the accuracy of random forest models to classify individuals based on either westernize/non-westernized, healthy/diseased, or male/female with either tree-agnostic diversity measures (e.g., the Shannon index) or with tree-based measures (e.g., Faith’s PD). The models that included tree-based measures were MUCH more accurate than the tree-agnostic models!
The inclusion of tree-based diversity measures in shotgun metagenomics is just the start in terms of integrating genomic information into studies of microbial diversity. For instance, many other measures of “function” can be used, and there are diversity measures that more explicitly incorporate function, versus our main approach of creating “functional similarity trees”. However, most existing algorithms for calculating functional diversity do not scale well to 100’s or 1000’s of functions across 1000’s of species. In addition, there are many exciting possibilities for integrating phylogeny and function more directly into machine learning models, such as via embedding approaches (e.g., word2vec or node2vec) that are often used in deep learning.
In summary, the notion that “everything makes sense in the light of evolution” has often been overlooked when assessing diversity via shotgun metagenomics due to the challenges of inferring a phylogeny. However, the proliferation of reference genomes enables a rethinking of how researchers can explore microbial diversity. Our Environmental Microbiology publication lays some of the groundwork for the development of “genome-based diversity”.